Methods to measure major particle composition – Posts from former Forum (2011-2012)
On the table below you can read the posts of the forum on “Methods to measure major particle composition” which was maintained by Phoebe Lam in 2011-2012. This forum is not any more available.
|
Welcome–1st posting was created by Phoebe Lam, 08 Dec 2011 23:20
Welcome to all who are interested in measuring major particle composition (POC, CaCO3, opal, lithogenics). What: The purpose of this forum is to provide an interactive space to discuss people’s experience with various methods, and to provide a resource for those who haven’t made these measurements before. I have started the forum with a summary of the motivations for starting this forum, and then a summary of the methods for measuring major particle composition that I am aware of based on my experience, some literature review, and conversions with Jim Bishop, Mark Brzezinski, Rob Sherrell, and Steve Manganini. Note that this is not a formal or comprehensive review, but simply a starting point for discussion. Please read through the post, and add your comments about your experiences with these methods, or comments describing alternative methods together with their merits and liabilities. How: Reply to the post by clicking “Quick Reply” for a text-only reply, or “Reply” for more formatting functionality. Click on “Subscribe” if you wish to have yourself notified about replies to this topic. If you wish to start a new topic discussion, please click on “New Topic”. Why: One of the recommendations to emerge from the 3rd GEOTRACES Data-Model Synergy Workshop held 14-17 November 2011 in Barcelona, Spain, was the importance of measuring total suspended particulate mass (SPM) and major particle composition (particulate organic matter (POM), CaCO3, biogenic silica, and maybe oxyhydroxides) for GEOTRACES. Since major particulate phases are not always routinely measured by the GEOTRACES community, and since they were not part of the official GEOTRACES intercalibration activities, this forum is meant to be a resource to those new to these measurements, as well as provide a forum for discussing the advantages and disadvantages of methods currently in use. An email from Francois Lacan summarized it nicely: “In my opinion the Barcelona Geotraces data/model synergy workshop confirmed the high importance of measuring particle dry weights – or better, particulate TEI carrying phase concentrations- on GEOTRACES cruises. It seems to me that the modellers need that information (as they work with particle concentration fields), but that the experimentalists often don’t measure it (often experimentalists report their data as mass of TE per mass of water, because they don’t know the mass of particles per mass of water). I believe that this situation is an obstacle for data/model synergies.” There was general (but not unanimous) consensus that obtaining suspended particulate mass by the method of adding up the major chemical phases of particles (“chemical dry weight”) was more efficient than direct measurements of dry weight (“gravimetric”), as it circumvents the difficulties associated with adequately correcting for the mass of residual salt for gravimetric dry weights (eg. Lam and Bishop, 2007, Deep-Sea Research II, 54: 601-638 ), and at the same time provides major particle composition information. Jim Bishop showed that when done carefully, chemical and gravimetric dry weight had good agreement for open ocean size-fractionated particles (eg. Bishop et al. 1977 Deep-Sea Research, 24: 511-548 ). Summary of methods: Particulate organic matter (POM): CaCO3 / Particulate Inorganic Carbon (PIC): Biogenic silica/opal: Lithogenics: Oxyhydroxides (may be important for hydrothermal vent plumes): *** |
|
Replied by Aristomenis P. KARAGEORGIS on topic Welcome–1st posting, 23 Dec 2011 20:08 During the Barcelona Geotraces meeting I asked why SPM concentration is not determined routinely by water filtration, and I realized that other colleagues, like Francois Lacan, had similar concerns.
I think the chemical determination of all (?) SPM constituents is too much trouble and involves many, and different types of analytical methods, as described in the literature cited by Phoebe. I don’t believe that all groups who need an SPM concentration have the ability to make all the analyses required. The major problem seems to be salt, which in the case of pumps cannot be removed. However, I have a relatively simple on-board filtration setup, which can eliminate the salt problem. The idea is to filter e.g. 10 L of seawater using light (e.g. polycarbonate filters; 47mm diameter, 0.4 micrometers pore-size) and be very careful to rinse several times the filter with MilliQ (pH adjusted) to remove sea salts before they crystalize. Essentially that means you have to be in the lab many hours and wait until filtration is about to end, or to decide at which volume you will stop filtration. Sea salts can be easily removed while in solution. We have seen that in scanning electron microscope images, where some filters are completely salt-free, and in others salt cubes are abundant. If someone gets tired and goes to bed, leaving the filter to dry, can explain the latter situation. The rest of the procedure is more or less known. Weighing the filters very carefully prior and after filtration is crucial. The balance room and equipment must be ideal and if possible the personnel performing measurements should be the same person. An estimate of particulate matter concentration can be also correlated against transmissometer measurements (beam c), and a marked correlation can provide much more useful data. Likewise, particulate organic carbon concentration can be correlated against beam c. I strongly encourage Geotraces colleagues to measure routinely particulate matter concentration because it is a fundamental parameter that can be used in many applications, and modelling in particular. I will be glad to provide any details regarding the filtration set-up, and some results can be found in Karageorgis et al., 2008. Deep-Sea Res. I, 55: 177-202 . Aris Karageorgis |
|
Replied by Phoebe Lam on topic Welcome–1st posting, 24 Jan 2012 20:07 Dear Aris,
Thanks for your posting and to the reference you provided. Your suggestion may be useful for some colleagues as an alternative to measuring SPM directly if chemical determination of major SPM constituents is impossible. One of the problems with weighing filters as you suggest is that particle collection, at least on the US GEOTRACES cruises, has moved to the use of depth filters such as quartz fiber filters (QMA) and polyethersulfone (Supor) filters. This is because these depth filters have far superior flow characteristics than polycarbonate filters, allowing in-situ pump filtration of the much larger volumes that are necessary for measurement of some radionuclides (through QMA and Supor) and also faster filtration times from in-line filtration from GO-Flo bottles (through Supor). The trade-off for better flow characteristics is more seawater retention by these filters, and thus more salt, making weighing impractical on the particle samples that are already being collected for TEI measurements. Measuring SPM as you suggest would require a dedicated person at sea as well as a dedicated bottle for this single measurement, which may be difficult with the limited berths and water budgets that have characterized the US GEOTRACES cruises, but may be possible for other cruises. Cheers, Phoebe |
|
Replied by Phoebe Lam on topic Welcome–1st posting, 11 Feb 2012 03:16 I received an email regarding biogenic Si analysis from Sven Kretschmer of AWI on February 1, 2012. I’ve copied his email as well as my response. —
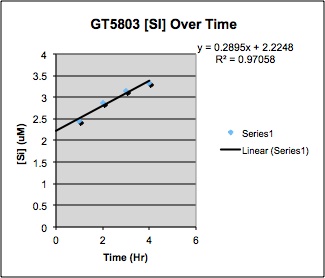
I will partly adopt your time series protocol and now that I will start my first test soon, I would like to kindly ask you some more questions regarding this protocol:Is the 0.2molar NaOH not too strong? I am afraid that the reaction goes to fast. On what it depends if you take the subsamples every hour or every half hour? I still don´t understand quite well how to calculate the bSi from 4 timepoints. Aren´t these too few points to make an extrapolation? Are you able to calculate the lithogenic Si from these 4 timepoints, or is there a need to make a final leach with weak HF in order to know the total Si? Is it of interest at all to know the lSi, because I will anyway determine the lithogenic component by another (multielemental) analysis. When measuring on the spectrophotometer why is the transmission more accurate than the absorbance?– I’ve pasted below what one of our timeseries looked like using 0.2M NaOH. We took samples at 1, 2, 3,4 hrs, then fit a line. The interpretation is that biogenic silica comes off quickly (before the first time point), but lithogenic silica dissolves slowly and linearly with time (see Demaster 1981). The intercept (in this case 2.2uM) was taken to be the biogenic silica. … [We are] going to do some tests to compare the slopes using 0.2M NaOH and 1M Na2CO3, and see whether we can get away with doing fewer time points.You cannot get lithogenic Si from these timepoints only; you would have to do a final leach with HF to get that. I skip that part, and calculate lithogenic material using Al in my ICP-MS runs of a HF-HCl-HNO3 digest.Re transmission vs absorbance: I think the idea is that the instrument measures transmission, whereas absorbance is calculated using measured transmission in a reference, your sample, and in the dark. If tehre is drift in the reference or dark, then the calculated absorbance will be off. We measure the reference and dark regularly to monitor for drift and calculate absorbance.
 |
| Replied by Sven Kretschmer on topic Welcome–1st posting, 16 May 2012 10:27
Hi Phoebe, |
| Replied by Phoebe Lam on topic Welcome–1st posting, 16 May 2012 12:27
Hi Sven, |
| Replied by Sven Kretschmer on topic Welcome–1st posting, 16 May 2012 13:00
Hi Phoebe, |